

View on GitHub
Open this notebook in GitHub to run it yourself
Quantum Thermal State Preparation Algorithm Implementation
Introduction
This implementation is based on the paper [1] and was written in collaboration with Chi-Fang (Anthony) Chen, the first author of the paper. Quantum thermal state preparation is the task of preparing the Gibbs state [2], which is the quantum state in a thermal equilibrium. In difference with ground state preparation, here one looks for properties of a quantum system which is coupled to an environment in a certain temperature. Besides simulating nature’s behaviour, it is an algorithmic primitive that can be used within other quantum algorithms, such as for solving semi-definitie programs [3]. Formaly, the Gibbs state is a mixed-state, and defined as: Notice that since the state is a mixed-state, we describe using a density matrix.Problem Definition
-
Input:
- : Hamiltonian of a system; Can be a pauli decomposition, block encoding or a function that efficiently implements .
- : Inverse-temperature of the system.
- Output:
- A state which is an -approximation of the Gibbs state: where denotes the trace distance.
Algorithm Description
The algorithm is a quantum version of the classical Markov chain Monte Carlo (MCMC) algorithm [4]. It is composed of the following steps:- Apply a random jump\transformation on the state from a set of jumps (usually a local transformation, but not necessarily).
- Measure the energy difference .
- Accept the jump with probability that is propotional to , otherwise reject the step and revert to the original state.
Challenges
There are 2 main challenges in translating the classical monte carlo to a quantum version:- Energy uncertainty: measuring the energy with high resolution is with exponential cost due to energy-time uncertainty.
- Rejection step: it turns out that rejecting is not trivial, as the accept involves measurements.
Running Time
The total simulation time will be , where is the mixing time of the Linbladian, measuring the time it takes to different mixed states to be indistinguishable under the evolution of . The mixing time is not trivial to calculate, and can be estimated in certain cases, such as [8]. The algorithm might give exponential advantage for quantum systems that thermalize fast enough and have the sign problem [9] so that no efficient classical alternatives exist.Algorithm implementation with Classiq
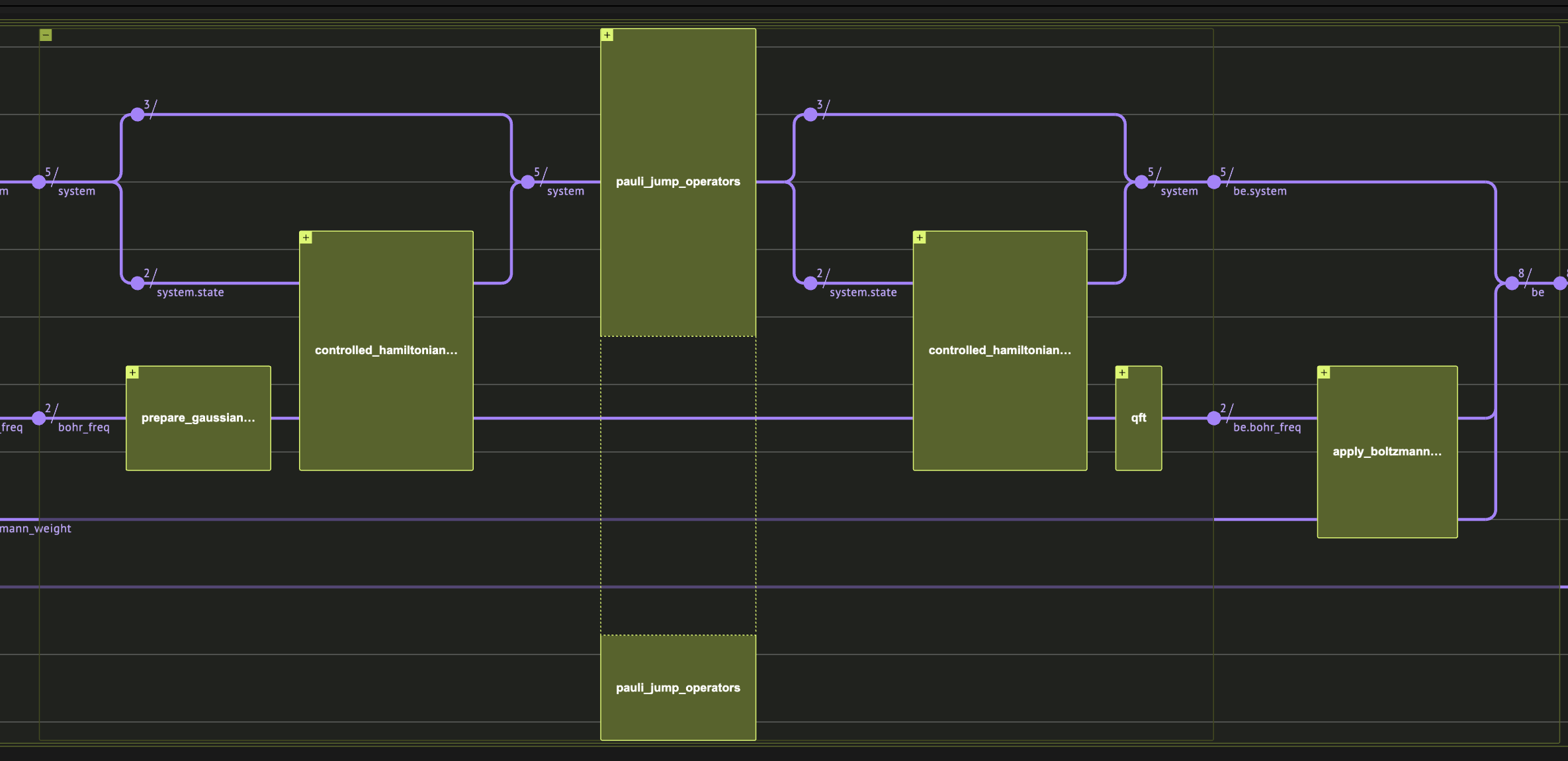
Toy problem setting
Here we define a very basic problem with just 2 qubits, and a diagonal hamiltonian, so that we will be able to implement the hamiltonian simulation exactly, and get results within the quantum simulator limits. We note that the algorithm does not gurantee improvement over classical monte carlo in the case of such hamiltonian, and we choose it for a didactic reason. The hamiltonian will be H = Z_1 Z_2 + Z_1 I = \begin\{pmatrix\} 2 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 \\ 0 & 0 & -2 & 0 \\ 0 & 0 & 0 & 0 \end\{pmatrix\}. The second parameter for the problem is . Here we choose quite small value for fast convergence.Operator Fourier Transform
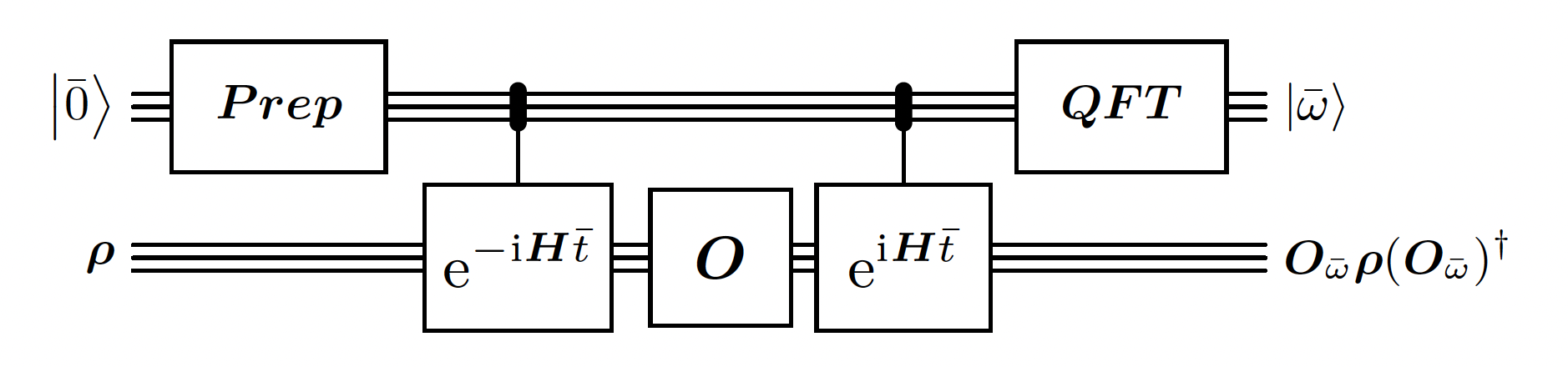
This is the heart of the algorithm. This building block is quite similar to the Quantum Phase Estimation. However, there are 2 main differences:- The initial state prepared for the phase register is a Gaussian distribution instead of a uniform one. It is used in order to have better guarantees on the error of the estimated energy. It is required because the time window is limited.
- The circuit is measuring the energy difference that the operator is doing on eigenvalues instead of just the energy of each eigenvalue in superposition.
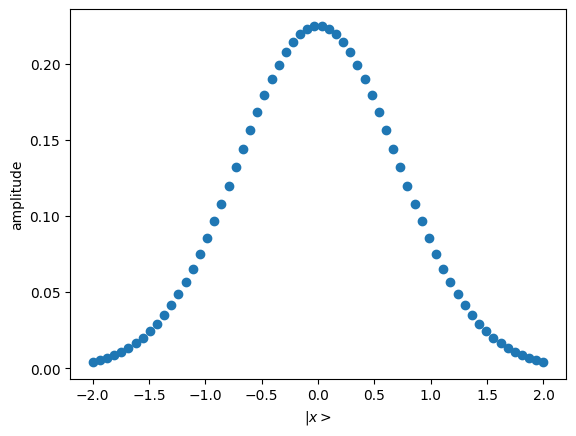
- First we define a gaussian state preparation function. We choose truncation value and in a quite arbitrary way now, qualitatively such that the gaussian trend will be within our truncation.
bohr_freq as signed integer, bohr_freq and bohr_freq,
while . It is analogous to the phase variable in Quantum Phase Estimation.
Require that , so that the energy estimation won’t overflow. We also want to take advantage of the full resolution of the QFT, so we choose close to the bound.
Output:
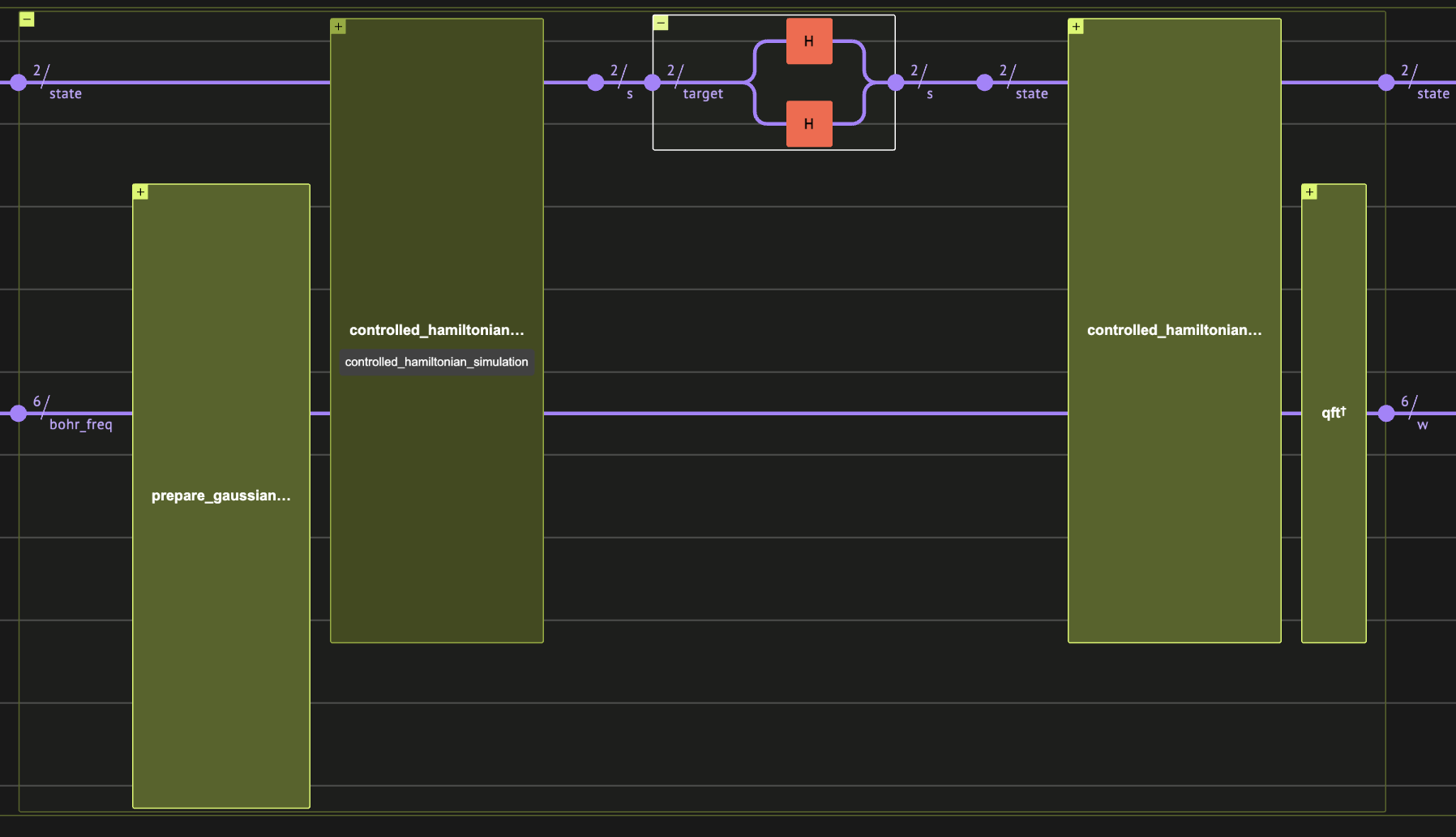
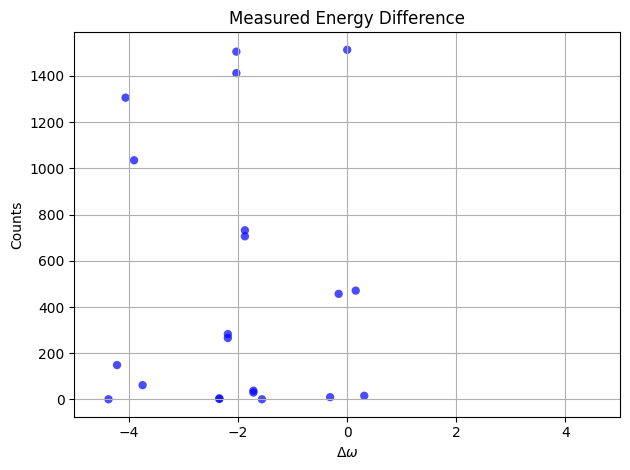
bohr_freq:
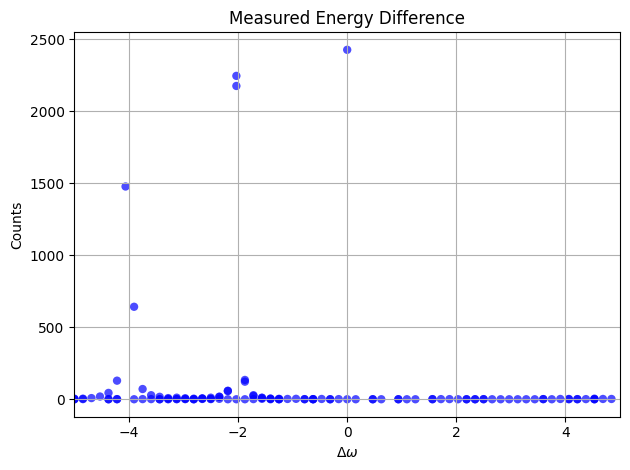
Linbladian Block Encoding
First we pick jump operators that will define our Linbladian dissipative part. It is enough to choose local operators, so here we just use all possible single-site pauli operators.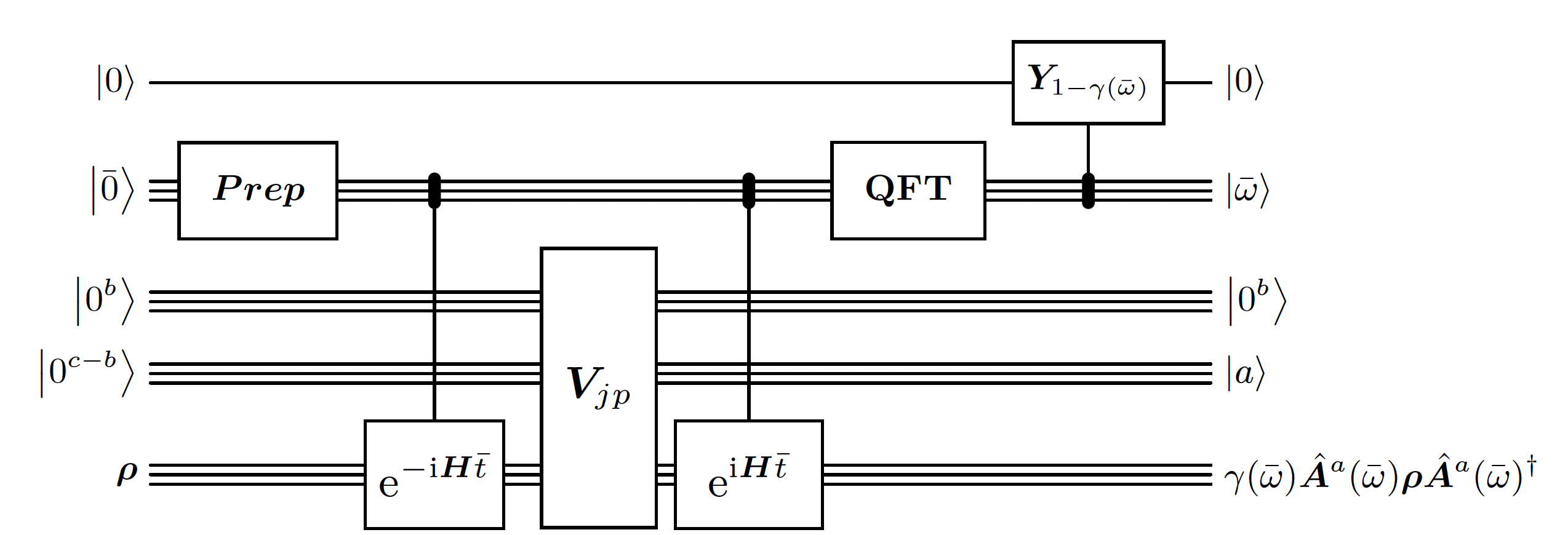
Linbladian Evolution of a -time step
Given our block encoding , and assuming that the Linbladian is purely irreversible, it is possible to evolve it for a timestep using a 1st order approximation: using weak measurements (weak measurement is a measurement that reveals only small amount of information on the system, and does not collapse entirely the state). Actually the it exploits a quantum Zeno-like effect [7] that makes the corrections to the evolution on quadratic in ! Note: This method is the 1st order approximation of the evolution. There are better scaling methods with higher order approximation to simulate the evolution of the Linbladian (see Appendix. F in the [1]).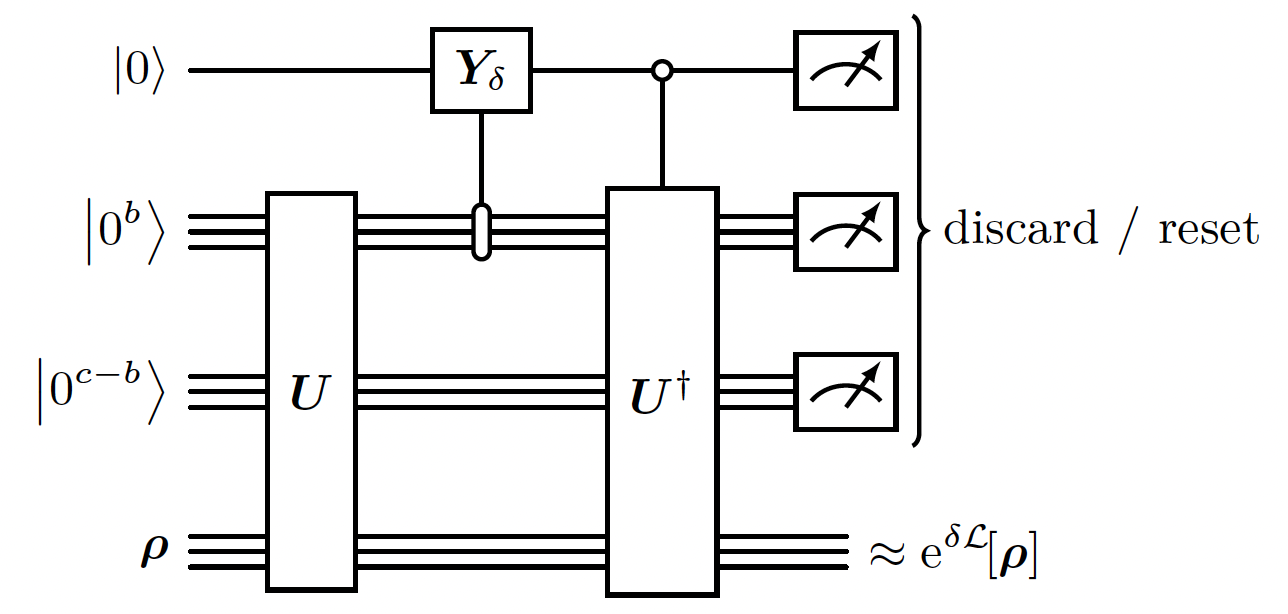
- We use the
RESETfunction to reset the state of a single qubit (thus saving qubits).
Running The algorithm
We choose parameters for the energy evaluation in the operator Fourier transform, a small enough delta timestep and number of repetitions. Note that as we makeDELTA small enough, the approximation of evolution will be better. As the MAX_REPETIOTIONS * DELTA (total simulation time) is larger, the state is better mixed and we get closer to the approximated detailed balance state.
As FT_WINDOW_SIZE is larger, the approximated detailed balance state gets closer to the desired detailed balance state.
Here we choose parameters that will allow reasonable simulation time on a quantum simulator.
Note that as the algorithm includes mid-circuit measurements, the runtime time of state-vector simulators scales linearly with the number of shots. If a high number of shots is required, it might be beneficial to use the density matrix simulator instead.
Output:
Output:
Output:
Output:
Notes:
- Exact detailed balance: astonishingly, in a followup paper [10] the authors improved the algorithm to reach exact detailed balance instead of approximated, still with finite time hamiltonian simulation.
- **Block Encoding vs.
- Coherent version: the paper also presents a purified version of the algorithm, that improves quadratically the run time.